
From Gut to Brain: A Fast Track to Memory Loss
Maybe "you are what you eat" wasn't so crazy after all.

A new mouse study suggests some memory decline may begin in the gut—not the brain—with inflammation disrupting communication along the gut-to-brain pathway. This same inflammatory pathway may help explain why GLP-1 drugs like Ozempic are associated with a lower risk of dementia, even though major human trials have failed to slow cognitive decline in people already suffering from Alzheimer’s disease. To me, the most important takeaway is that brain aging may be more modifiable than we assume. Rather than focusing only on downstream symptoms after cognitive decline has emerged, this research points upstream—toward earlier biological drivers such as inflammation, metabolism, and gut-to-brain signaling.
Here it is! My inaugural Field Note. After 25 years in cybersecurity analysis—learning to explain upstream risks before they became crises—I’m now applying that same mindset to brain health. I’ve often written for clients about “shifting left” to find vulnerabilities at the source before they reach production. Brain health needs the same rethink: many current treatments target downstream effects such as low serotonin levels and amyloid plaques rather than root causes. We need to focus upstream!
In this first Field Note, I’m writing about a new study in mice that links gut bacteria to memory loss, and potential ways to reverse that loss. BTW, my interest in the gut-to-brain connection started a few years ago after reading about a patient who developed depression following a poop transplant (it’s a thing!) from a donor with undisclosed depression.
If these mouse study results prove relevant to humans, treatment strategies for cognitive decline could fundamentally change. Below, I explain why understanding this pathway is essential.

“What we call ‘brain aging’ may partly stem from a breakdown in gut-to-brain communication.”
Age-related memory decline is top of mind. My family history makes me extra sensitive. For example, recently, when I couldn’t immediately recall the name of the Canadian singer who went to school with my Uncle (Leonard Cohen), I freaked! Friends told me, “Happens to me all the time.” But why do our memories fade and become harder to reach with age?
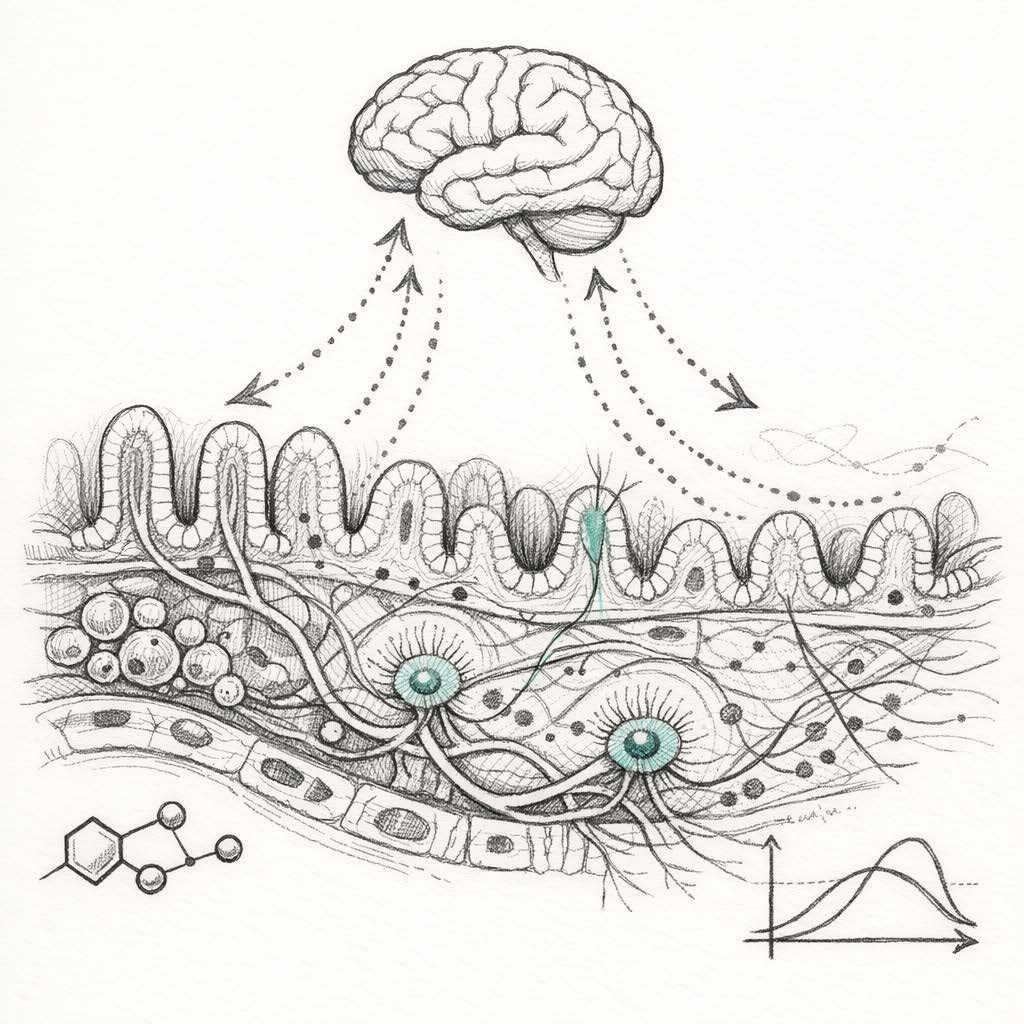
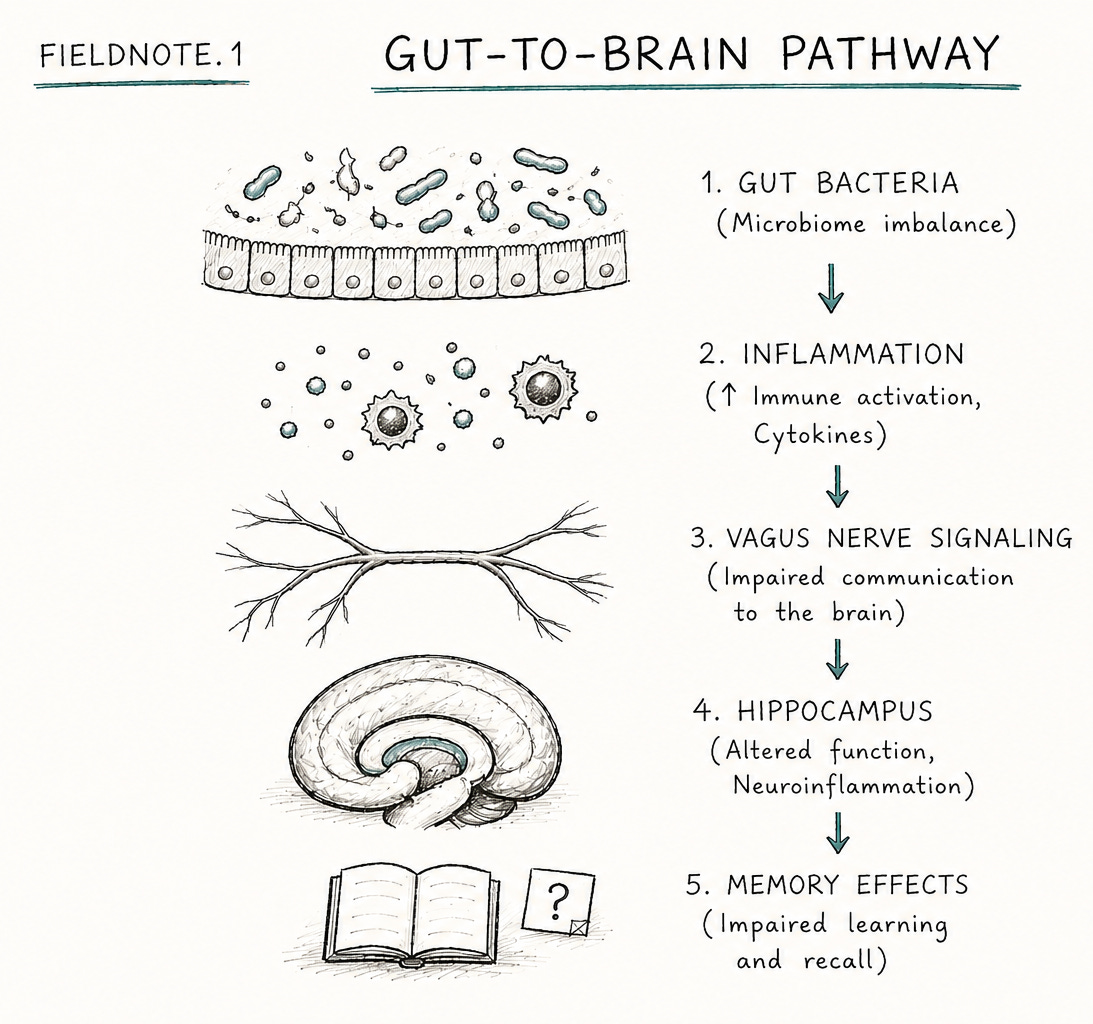
Researchers at the University of Pennsylvania suggest that what we call “brain aging” may partly stem from a breakdown in gut-to-brain communication. The gut bacterium Parabacteroides goldsteinii in mice produces medium-chain fatty acids. These activate receptors on peripheral immune cells, triggering inflammation. This inflammation damages vagal nerve impulses to the hippocampus (where new memories are made).
What’s intriguing is that this finding is consistent with another area of research in humans: peripheral inflammation is linked to cognitive decline, and may cause it. Two large meta-analyses found that higher levels of inflammatory markers significantly increase the risk of cognitive decline in older adults. Even more striking, a long-term study found that people with higher inflammation in their 20s performed worse on cognitive tests in middle age. However, while these studies suggest an association, they do not confirm causation or the exact mechanisms involved. Still, inflammation is increasingly hard to ignore.
What excites me about this paper is the level of detail in the biological pathway. This isn’t just generic advice about “gut health” — it’s a description of a specific molecular pathway.
The researchers tackled causation versus correlation head-on using multiple approaches. Young mice developed memory problems after exposure to aged mice and their gut bacteria. Germ-free mice receiving microbiomes from older mice showed a similar decline. Researchers also isolated specific bacterial species to test their direct effects.
Researchers linked cognitive decline to reduced activation of hippocampal neurons. They identified medium-chain fatty acids as an active molecule that could reproduce memory deficits when given directly.
Further, they tested specific interventions. A bacteriophage targeting bacteria improved memory in aged mice. Inhibiting a specific cell-surface receptor involved in inflammation triggered by the medium-chain fatty acids restored cognitive function. Finally, Vagal nerve stimulation with capsaicin restored memory performance.
The mouse research is quite interesting to me because it relates to drugs that millions of people already take. Researchers showed that liraglutide (similar to Ozempic and Wegovy) improved memory in aged mice. This finding may relate to human findings. Strikingly, observational studies have linked GLP-1 drugs for diabetes to lower dementia risk. Impressive, right, though this may be correlation more than causation.
Late last year, Novo Nordisk announced that its EVOKE trials, the largest GLP-1 studies conducted in Alzheimer’s, did not slow cognitive decline, prompting Novo to end the program. This doesn’t kill the GLP-1 hypothesis, but it complicates it significantly.
“Inflammation, metabolism, and neurodegeneration may be parts of the same upstream process.”
I’m intrigued by the way GLP-1 drugs sit at the intersection of metabolism, inflammation, and cognitive decline. Researchers increasingly suspect that insulin resistance, impaired glucose metabolism, and chronic inflammation may all contribute to cognitive decline and Alzheimer’s disease. This is one reason some researchers describe aspects of Alzheimer’s as “Type III diabetes,” since many Alzheimer’s brains show disrupted glucose utilization.
It’s important to note that the observational studies tracked people taking GLP-1 drugs before dementia onset. The EVOKE trials tested the drugs in people who already had early-stage Alzheimer’s. In other words, the drugs may still have value for prevention even though they don’t reverse existing damage. This difference—between prevention and treatment, and between causation and correlation—highlights a research gap that will require more targeted trials to resolve.
What makes this worth paying attention to is that brain aging may be driven in part by modifiable factors. Aged, germ-free mice experience delayed cognitive decline. This makes the upstream focus personally urgent. And by targeting defined actors along the pathway, researchers suggest a possible route to truly tackle the causes—not just the symptoms—of cognitive decline, at least in mice. Interestingly, the researchers did not find increased intestinal permeability (“leaky gut”) in the mice experiencing cognitive decline, suggesting the effects may be more closely tied to inflammation and vagal signaling.
How cool is it that specific approaches can restore memory function in aged mice? This could open a path toward new treatments targeting inflammation and gut-to-brain pathways in humans.
“Mickey ain’t me!”
Despite all the Facebook videos we see, plausible isn’t the same as proven. This study shows a clear mechanism in mice, but at the same time, Mickey ain’t me! Whether this gut-brain pathway is the same in humans, whether bacterial changes occur similarly across species, and whether the tested interventions would prove safe and effective in people are the essential unknowns I will follow closely.

GLP-1 and dementia meta-analysis — 26 trials, 160,000+ patients
Early inflammation and cognitive decline — Yaffe/UCSF, Neurology 2024
IL-6 and cognitive decline meta-analysis — 15,828 participants

I’m intrigued by the possibility that the gut-to-brain pathway has an impact beyond memory and cognition. This investigation will continue as I explore the vagus nerve’s role in treatment-resistant depression. If it turns out that high inflammation and vagal signaling suppression contribute to both cognitive decline and mood disorders, these pathways may prove more relevant than we currently understand.
If you’re not a subscriber, please subscribe to get full access to the newsletter and publication archives.